
In the dynamic and highly regulated pharmaceutical industry, achieving regulatory approval is not just about developing a quality product it’s about proving its safety, efficacy, and equivalence with precision. Among all the essential components of a regulatory dossier, the Bioequivalence (BE) Study holds exceptional significance. It serves as the scientific bridge that assures regulatory authorities your formulation performs the same as the reference product.
Yet, despite its importance, many companies face repeated hurdles during regulatory submissions. Surprisingly, these failures often stem not from product issues, but from incomplete BE data, poor documentation, unverified CRO processes, or lack of regulatory alignment. As agencies worldwide tighten their standards, even minor deficiencies can lead to long delays, rejections, or costly resubmissions.
Bioequivalence (BE) Studies are a cornerstone of global dossier submissions. Regulatory agencies rely heavily on BE data to ensure that a generic formulation matches the safety, efficacy, and PK profile of the innovator product. However, despite their importance, a significant number of BE studies and consequently dossier submissions fail due to avoidable gaps in planning, documentation, CRO compliance, and data integrity.
With regulatory expectations becoming more stringent and inspection frameworks more thorough, companies must ensure their BE studies follow global best practices and comply with the guidelines issued by major authorities such as US FDA, EMA, WHO, CDSCO, ICH, and others.
Why Bioequivalence Study for Dossier Submission Fails: Key Challenges
Many BE studies fail because of lapses that occur long before data reaches regulatory authorities. Companies often face challenges due to poor scientific planning at the protocol stage, such as choosing an incorrect study design (fasting/fed), wrong reference product, or misaligned sampling time points issues clearly outlined in guidelines such as the FDA’s Bioequivalence Guidance and the EMA’s Guideline on the Investigation of Bioequivalence.
Another frequent cause of failure is insufficient documentation, including missing raw bioanalytical data, incomplete method validation, deviation logs, or inconsistent audit trails. These documentation gaps are widely recognized in regulatory references like FDA BIMO inspection findings and ICH M10 (Bioanalytical Method Validation).
CRO non-compliance also plays a major role. Agencies like WHO and ANVISA emphasize data integrity and adherence to GCP/GLP. When CROs fail to follow proper SOPs, maintain QC/QA records, or justify deviations, the integrity of the entire study becomes questionable.
Weak coordination between the sponsor and CRO results in delays, unclear communication, and missing documents. When regulatory authorities raise queries, companies often struggle to respond quickly and comprehensively because the CRO does not provide timely support something repeatedly highlighted in global inspection trends and published surveys on BE submission deficiencies.
Finally, many studies fail during audits or data verification due to inconsistencies in chromatograms, sample handling, or protocol deviations. The FDA’s “For-Cause Inspections in BE Studies” and WHO’s Handbook on Bioequivalence Testing identify these issues as major contributors to failed submissions.
Reasons
1. Insufficient Scientific Planning During Study Initiation A successful BE study begins with strong scientific groundwork. Many failures occur because the foundational elements are not planned in alignment with regulatory expectations. When the initial design lacks scientific rationale, the entire study becomes vulnerable. Common issues include selecting an inappropriate study design, misjudging sampling intervals, choosing unsuitable reference products, or defining inclusion/exclusion criteria that do not meet regulatory standards. When these early decisions are not scientifically sound, authorities often question the overall reliability and acceptability of the study outcomes.
2. Incomplete or Weak BE Study Documentation Regulatory bodies depend heavily on accurate and comprehensive documentation to verify the credibility of a BE study. Submissions are frequently delayed or rejected due to gaps such as missing raw bioanalytical data, incomplete method validation reports, absence of audit trails, unsigned consent forms, or unclear statistical justifications for subject exclusions. Even minor inconsistencies in documentation can raise red flags, leading to extensive regulatory queries and prolonged evaluation cycles. Thorough, transparent documentation is essential to demonstrate compliance and ensure smooth review.
3. CRO Non-Compliance and Inadequate Audit Readiness Another major reason for BE study failures is the selection of a CRO that does not fully comply with international regulatory standards. Regulatory agencies—including WHO, FDA, EMA, GCC, and ANVISA—conduct detailed inspections and expect CROs to maintain robust practices. Problems arise when CROs fall short in areas such as GCP/GLP compliance, data integrity controls, deviation handling, or bioanalytical method validation. Any weakness detected during a regulatory review undermines confidence in the study, putting the entire submission at significant risk.
4. Ineffective Coordination Between Sponsor and CRO A BE study’s success depends heavily on continuous communication and mutual clarity. When coordination is weak, sponsors often experience delays in updates, incomplete responses to queries, and last-minute documentation gaps that disrupt submission timelines. Misalignment also occurs when regulatory instructions are misunderstood or not communicated promptly. These communication lapses lead to avoidable complications, making the final dossier less cohesive and more susceptible to regulatory scrutiny.
5. Slow or Poorly Structured Responses to Regulatory Queries Timely and well-supported responses are crucial once authorities raise queries. Many submissions fail because responses are delayed or lack the necessary scientific justification and supporting evidence. In some cases, CROs do not provide clear explanations or fail to conduct a proper root cause analysis. Weak query management not only affects credibility but also significantly reduces the likelihood of obtaining approval during the first review cycle.
6. Data Integrity Concerns and Analytical Inconsistencies Regulators are increasingly vigilant regarding data integrity. Any inconsistency—whether in chromatographic patterns, sample handling logs, traceability records, statistical evaluations, or protocol adherence—raises serious concerns. Even unintentional lapses may signal deeper issues in data management and quality control. When integrity is questioned, the submission faces substantial delays or outright rejection.
Real-World Case Examples
Here are a few anonymized examples that reflect common industry situations:
Case 1: Rejection Due to Incomplete Raw Data
A company completed its BE study successfully, but the authority rejected the dossier because the CRO failed to maintain electronic audit trails for chromatographic data violating USFDA data integrity requirements.
Case 2: Protocol Deviation Not Reported
Another sponsor faced delays because the CRO did not document minor protocol deviations. During audit review, the authority noted missing logs, raising concerns about data reliability.
Case 3: Study Accepted After Corrective Actions
A third company faced issues during WHO PQ evaluation due to incomplete AMV records. After collaborating with an experienced regulatory partner, corrective actions were implemented, documentation gaps were fixed, and the dossier was eventually approved.
What Companies Should Do? A Practical Checklist
Before Starting the Study
- Map regulatory requirements based on target country guidelines.
- Select CROs with proven audit history (USFDA/MHRA/EMA/CDSCO).
- Verify analytical method validation SOPs and capabilities.
- Ensure protocol is aligned with the latest guidelines (EMA 2010, USFDA 2018, WHO TRS 1003).
During the Study
- Maintain continuous communication between sponsor, CRO, and BA/BE teams.
- Ensure sample handling, storage, and bioanalysis follow ALCOA+.
- Monitor protocol deviations and document them properly.
- Conduct unannounced internal QA checks.
Before Submission
- Perform pre-audit verification of full raw data.
- Review AMV, PK analysis, and study report alignment.
- Conduct a mock regulatory audit.
- Compile documents as per CTD/ACTD/eCTD structure.
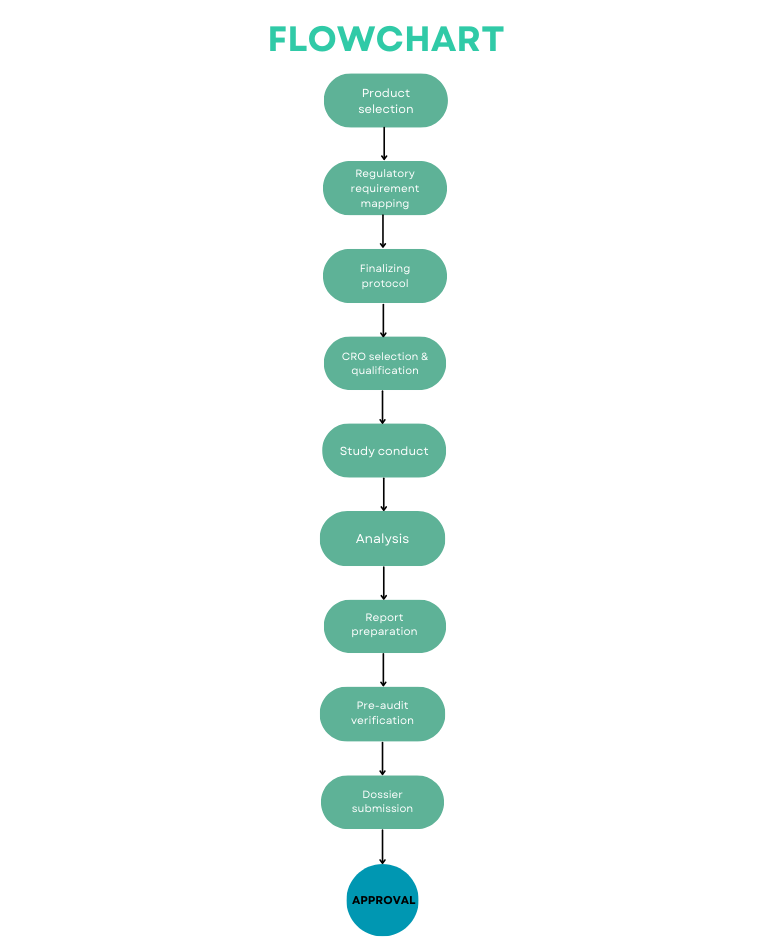
Simple BE Study Process Flowchart
Common Mistakes Companies Make
- Choosing a CRO based only on low cost instead of compliance history.
- Using incomplete AMV reports or outdated SOPs.
- Not performing pilot studies for complex or variable drugs.
- Ignoring regulatory guideline updates.
- Not conducting pre-audit data reviews.
- Relying solely on CRO without sponsor-side quality oversight.
- Submitting BE data without verifying bioanalytical raw data.
These mistakes often lead to authority queries, audit failures, or outright BE study rejection.
Future Trends in BE Studies
- Increasing focus on data integrity (ALCOA+ and 21 CFR Part 11 compliance).
- Growing use of replicate design for highly variable drugs.
- Higher demand for CROs with proven international audit readiness.
- Adoption of electronic source documentation and digital sample tracking.
- Rising expectations for detailed PK modeling and statistical justification.
- More stringent subject safety & ethical compliance requirements.
- Regulatory push for faster, more transparent dossier reviews globally.
How Partnering With Us Helps You Overcome These Challenges
Our approach ensures your BE study is designed, executed, documented, and presented in full compliance with the guidelines and best practices outlined above.
Conclusion
A BE Study is much more than a regulatory requirement it is a scientific and compliance-driven process that directly determines the success of your global submissions. Most BE study failures are preventable with proper planning, adherence to international guidelines, and collaboration with trusted CRO partners who maintain high-quality standards.
By working with us, you gain a partner who ensures your BE studies are scientifically sound, compliant with global guidelines, fully documented, and audit-ready leading to smoother evaluations and faster regulatory approvals.
At Regcure Pharma, we support companies with complete BE study planning, CRO qualification, documentation review, and pre-audit verification to help you achieve smoother submissions and faster approvals.
Connect with us:



